All published articles of this journal are available on ScienceDirect.

A Case Report: Type II Abernethy Malformation Complicated with Congenital Polydactyly and Enlargement of all Cardiac Chambers
Abstract
Background:
Abernethy malformation is a kind of congenital malformation of the portal vein system caused by abnormal portacaval shunts. It can be in combination with many other congenital malformations. There has been a limited number of patients since the first patient was reported, leading to limited knowledge of this kind of disease.
Methods:
In August 2018, we treated a patient diagnosed with type II Abernethy malformation complicated with both congenital polydactyly and enlargement of all cardiac chambers, which is extremely rare and can be supplementary to the existing cases. According to a comprehensive and adequate assessment of patients' condition, we treated him with oral silybin (70 mg every time and 3 times a day) for 3 months, and advised him to make follow-up visits.
Results:
At the latest follow-up, we knew the health condition of this patient was generally satisfactory, whether in terms of laboratory test results or his daily life experience.
Conclusion:
Although the major therapy for Abernethy malformation is surgery, this case suggests that simple conservative treatment with regular follow-up visits can be suitable for certain patients.
1. INTRODUCTION
Abernethy malformation, a rare hepatic vascular malformation, is a kind of congenital malformation of portal vein system caused by abnormal portacaval shunts. It can be divided into two types [1, 2]. Type I, to which many female and pediatric patients belong, is characterized by the absence of intrahepatic portal vein branches and an end-to-side portocaval shunt, patients in this type usually have a short medical history as a result of the rapid progress like liver failure, whereas in type II the intrahepatic veins are hypoplastic but patent and a side-to side shunt diverts blood from the portal vein to the Inferior Vena Cava (IVC) [3]. Abernethy malformation can be in combination with other congenital malformations such as cardiovascular malformations, biliary atresia, choledochus cysts, polysplenia or musculoskeletal abnormalities [3, 4]. Besides, patients may also suffer from some complications, including hepatic encephalopathy [5], hepatopulmonary syndrome [6], pulmonary arterial hypertension [7], even hepatocellular carcinomas [8]. For the reason that there have been only about 300 patients reported in the literature since the first patient was reported by Abernethy in 1797 [9] and the vast majority of these publications are single case reports, it is difficult to calculate the accurate prevalence of this disease.
2. CASE REPORT
We treated a patient in August 2018. This patient was diagnosed as type II Abernethy malformation (ICD-10 code: Q26.501) complicated with congenital polydactyly (ICD-10 code: Q69.901; OMIM code: #603596) and enlargement of all cardiac chambers (ICD-10 code: I51.709; OMIM code: #600001), which is extremely rare and can be supplementary to the existing cases. This patient was a 16-year-old boy with a 6-year history of low white blood cell (WBC) and platelet levels. Routine blood tests over the past 6 years showed that his WBC counts ranged from 2.36×109/L to 3.87×109/L and his platelets ranged from 48×109/L to 72×109/L, while those for erythrocytes and hemoglobin were normal. During the treatment that he received at his local hospital, the results of a bone marrow cytology test and bone marrow biopsy showed normal erythroid proliferation, while the granulocytic and megakaryocytic maturation were arrested. Fluorescent in situ hybridization (FISH) was used to detect the chromosomal or genetic abnormality. The probes for detection were D7S 486/CSP7 (7q31), EGR1/D5S23 and D5S721 (5q31), D20S108/CSP8 (20q12/CSP8), CEP8 SpectumAqua. The air-dry dropping method was used for chromosome preparation, and the working solution consisted of probe, water and hybridization buffer (the ratio of these three components was 1:2:7). Then the working solution was added to the hybridization area on slide glass substrates. The red / green or aqua fluorescence hybridization signals of interphase cells under excitation were observed by a fluorescence microscope. 200 bone marrow cells for total were analyzed. The results showed that 2 red and 2 green fluorescence hybridization signals or 2 aqua fluorescence hybridization signals were observed in each cell, which indicated neither -7/7q-, the absence of gene EGR1 (5q31) or D20S108, nor increased numbers of chromosome 8.
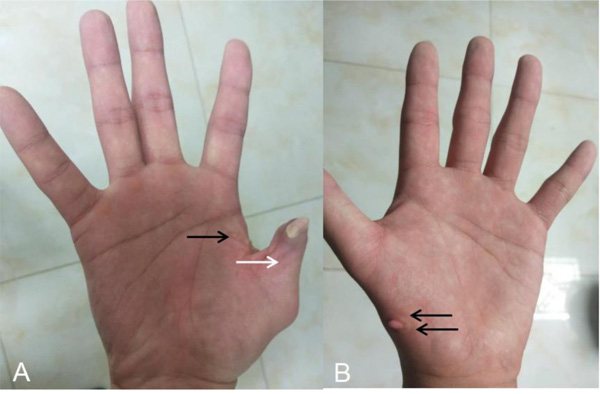
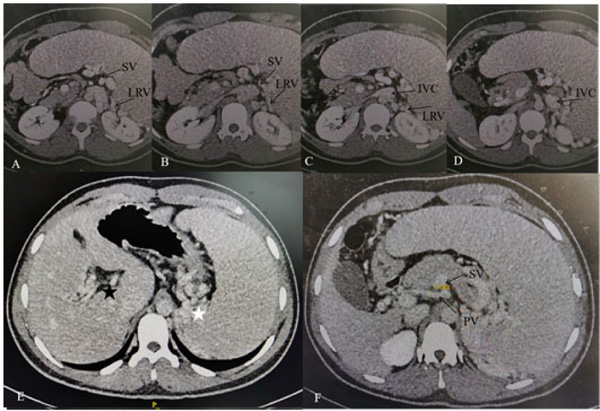
After he was admitted to our hospital, we knew there were no familial cases and no parental consanguinity. We gave him a basic physical examination and found his spleen was palpated at the bottom of the ribs with the second line 10.0 centimeters long, both of his hands had six fingers, and the surplus thumb on the right hand had been resected several years before, leaving a deformed and adducted one with ventrally displaced nail (Fig. 1A). A small and convex tissue was present on the medial surface of his left thumb, which was considered to be an incompletely developed finger (Fig. 1B). In addition, we gave some basic laboratory examinations for supplementary. The results were as follows: The levels of Alanine Aminotransferase (ALT) and Aspartate Transaminase (AST) were both normal, while that of direct bilirubin (11.2 umol/L) and indirect bilirubin (27.2 umol/L) were increased slightly. Prothrombin time (PT) was 15.60 seconds and prothrombin activity was 53.00 percent. Tests for serum GPIb and GPIIb/IIa platelet autoantibodies were positive. Antinuclear antibody, anti-dsDNA antibody, anti-SSA antibody, anti-SSB antibody, anti-RNP antibody, anti-SSA antibody, anti-Sm antibody, anti-CCP antibody, anti-Scl-70 antibody, anti-Jo-1 antibody were all negative. Abdominal ultrasound showed splenomegaly (ICD-10 code: D73.201) and cavernous transformation of the portal vein (CTPV) (OMIM code:601004), the shape, size and edge of his liver were almost normal. Computed Tomography Angiography (CTA) of the portal vein system showed CTPV (Fig. 2E), normal portal vein diameter (7 mm), splenomegaly, spleno left renal shunt (Fig. 2A to 2D) and spleno gastric shunt (Fig. 2F). For financial reasons, the patient refused to have an indirect portographic examination. The results of Ultrasonic Cardiogram (UCG) showed enlargements of all cardiac chambers, the diameter of his left atrium was 41 mm, right atrium 52 mm, left ventricle 55 mm, and right ventricle 26 mm. The Left Ventricular Ejection Fraction (LVEF) was 65 percent.
Taking these symptoms and examination results into consideration, we diagnosed the patient with type II Abernethy malformation complicated with congenital polydactyly and enlargement of all cardiac chambers. Because the patient’s liver function was mild disorder, cardiac function was normal despite the enlargement of all cardiac chambers, and no malformations or complications associated with other systems were found, we treated him with oral silybin (70 mg every time and 3 times a day) for 3 months, and advised him to make follow-up visits.
3. RESULTS
We attained at the latest follow-up that his platelets were 70×109/L and WBC were 3.81×109/L, he did not complain of any discomfort. Like his peers, he almost led to a normal life.
4. DISCUSSION
This case adds Abernethy malformation with both congenital polydactyly and enlargement of all cardiac chambers to that list. Besides, the case also illustrates the importance of making a differential diagnosis between Abernethy malformation and other diseases.
Abernethy malformation may lead to some changes of spleen in form and function, which can further bring hematological changes, and lead to a misdiagnosis as a few blood diseases such as Immune Thrombocytopenia (ITP) and Myelodysplastic Syndrome (MDS). In terms of this patient, making the differential diagnosis between Abernethy malformation and ITP, MDS, Systemic Lupus Erythematosus (SLE) should be taken into consideration. The reason why we need to make a differential diagnosis with ITP is his arrested megakaryocytic maturation and positive platelet autoantibodies. The points of differential diagnosis lie in the following two aspects. Firstly, ITP patients do not have splenomegaly or merely have mild splenomegaly. Secondly, the diagnosis of ITP demands the exclusion of other secondary factors that result in a decrease of platelets. According to the principle of monism, splenomegaly and hypersplenism secondary to Abernethy malformation can be used to explain the decrease of platelets. Therefore, we didn’t consider the diagnosis of ITP. The reason why we need to make a differential diagnosis with MDS is the decrease of his WBC and platelets. As we know, WBC and platelets may decrease in MDS patients, but the results of bone marrow biopsy in MDS patients always show excessive proliferation. However, the result of bone marrow biopsy in this patient showed arrested granulocytic and megakaryocytic maturation with normal erythroid proliferation. Besides, the result of FISH showed neither -7/7q-, the absence of gene EGR1 (5q31) or D20S108, nor increased numbers of chromosome 8 in this patient. Therefore, we didn’t consider the diagnosis of MDS. The reason why we need to make a differential diagnosis with SLE is that there was abnormal liver function, splenomegaly, arrested megakaryocytic maturation and positive platelet autoantibodies, which may also present in SLE patients. Nevertheless, according to the revised classification standard in 1997 from the American Rheumatic Association (ARA), this patient simply met one of the 11 criteria, which is a decrease of WBC and platelets. Therefore, we didn’t consider the diagnosis of SLE. In summary, when we treated patients with simultaneous peripheral blood cell decrease, blood diseases and rheumatic diseases should be attached to primary importance. However, for those patients combined with other congenital malformation and abnormal liver function, we also need to take Abernethy malformation into account when making a differential diagnosis. It is reasonable to give a CTA of the portal vein system, even indirect portographic examination, the result of which may be a meaningful guide for diagnosis.
CONCLUSION
In the existed literature, there was no standard therapy to treat Abernethy malformation [10], the major therapy of it included shunt closures through surgical or radiological interventions and liver transplantations [11]. A few case reports have described endovascular techniques for portocaval shunt closure [12-15]. In a retrospective study, 42 patients diagnosed with Abernethy malformation were treated with single operative ligation (n = 12), staged operative ligation (n = 18), endovascular occlusion (n = 8), combined surgical and endovascular closure (n = 2), and observation (n = 2). No significant difference was observed in the decrease of serum ammonia levels between patients treated surgically or endovascularly. For the patients treated with observation, no comparison was made due to the rarity of patients [16]. The experience in treating this patient also demonstrates that simple conservative treatment with regular follow-up visits can also be suitable for those patients who do not have severe complications, this depends on a comprehensive and adequate assessment of patients' condition.
LIST OF ABBREVIATIONS
| WBC | = White Blood Cell |
| FISH | = Fluorescent in Situ Hybridization |
| ALT | = Alanine Aminotransferase |
| AST | = Aspartate Transaminase |
| PT | = Prothrombin Time |
| CTPV | = Cavernous Transformation of the Portal Vein |
| CTA | = Computed Tomography Angiography |
| MPV | = Main Portal Vein |
| UCG | = Ultrasonic Cardiogram |
| LVEF | = Left Ventricular Ejection Fraction |
| ITP | = Immune Thrombocytopenia |
| SLE | = Systemic Lupus Erythematosus |
| MDS | = Myelodysplastic Syndrome |
| SV | = Splenic Vein |
| LRV | = Left Renal Vein |
| IVC | = Inferior Vena Cava |
| PV | = Portal Vein |
AUTHORS’ CONTRIBUTIONS
Conceptualization: Cheng Zhu.
Investigation: Cheng Zhu, Qian Hao.
Methodology: Cheng Zhu.
Project administration: Cheng Zhu.
Writing – original draft: Cheng Zhu.
Writing – review & editing: Min Wang.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The procedures followed were in accordance with the ethical standards of the committee on human experimentation (Qi-Lu Hospital of Shandong University, China), The present case report was approved by the Ethical Committee of Qi-Lu Hospital of Shandong University, China (Reference No: 2019197).
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
CONSENT FOR PUBLICATION
We have obtained a written consent-to-disclose form signed by the patient’s father.
CONFLICT OF INTEREST
The author declares no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We would like to thank the patient and his family, without whom this case report would not have been possible. We also thank our colleges in Qi-Lu hospital of Shandong university for
providing helpful support and advice on this case report.

