All published articles of this journal are available on ScienceDirect.

Association of Mannose-Binding Lectin Gene Polymorphisms with Liver Diseases: A Review
Abstract
Mannose-Binding Lectin (MBL) is a member of the collectin family and is an important protein in the immune system. It is a pathogen pattern-recognition molecule that binds to specific carbohydrate motifs on the surface of many pathogens. MBL activates complement via lectin pathway. Single nucleotide polymorphisms in the MBL gene influence serum MBL concentration and function. MBL deficiencies increase the risk of infection and disease-specific complications, especially in those who are already immune compromised with pre-existing conditions. This review discusses the molecular genetics of human MBL and the association of MBL polymorphisms with liver diseases including liver fibrosis, viral hepatitis B, viral hepatitis C, and infection post-liver transplantation.
1. MBL PROTEIN
The complement system, a major component of innate immunity, provides immediate defence against infection and has a pro-inflammatory response. It consists of plasma and membrane proteins, which mediate 3 pathways, namely classical, alternative and the most recently described lectin [1]. Activation of these pathways of cascading enzymatic reactions results in the deposition of fragments that promote inflammatory and immune response. The classical pathway is activated when Immunoglobulin M or Immunoglobulin G antibody bind to antigens such as viruses and bacteria that the resulting immune complexes are recognised by C1q, the first component of the classical pathway. The activation of lectin pathway is similar to the classical pathway except that pathogens are directly recognised by a lectin, which includes MBL. MBL is an equivalent of C1q.
Lectins are associated with MBL associated serine proteases (MASPs) that are structurally and functionally similar to C1q-associated proteases C1r and C1s. MASPs also cleave C2 and C4. The major effect of the complement system, regardless of the pathway, is the deposition of the complement fragment C3b on the target (Fig. 1). The effects are membrane modification and promotion of the inflammatory response. Complement fragments including C3b deposit in large numbers on microbes and unwanted materials such as apoptotic cells and necrotic tissue. This coating or opsonisation of bacteria and altered-self elements allows specific receptors on peripheral blood cells, especially phagocytes, to bind these ligands. Additionally, activation of complement results in the formation of the membrane attack complexes (C5b-9), which perturb the bacterial cell membrane, thereby causing lysis of the microbes. Activation of the complement system leads to the release of pro-inflammatory peptides. Additionally, complement is also bactericidal as C3a and C4a directly kill microorganisms [2].
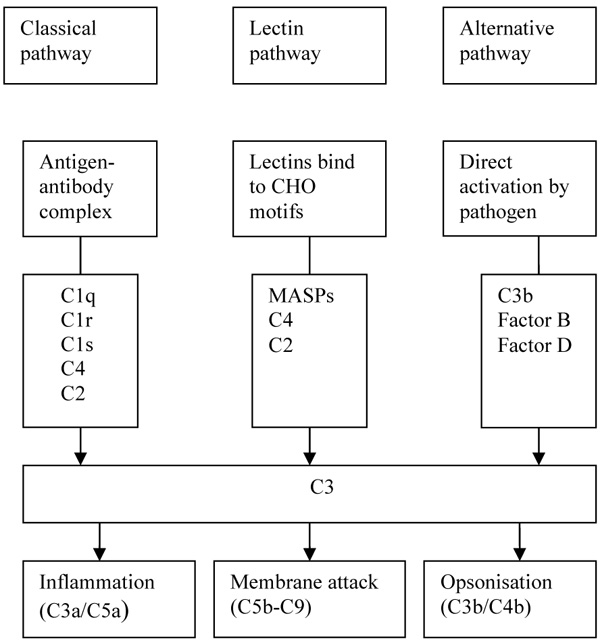
CHO: Carbohydrate, MASPs: MBL Associated Serine Proteases.
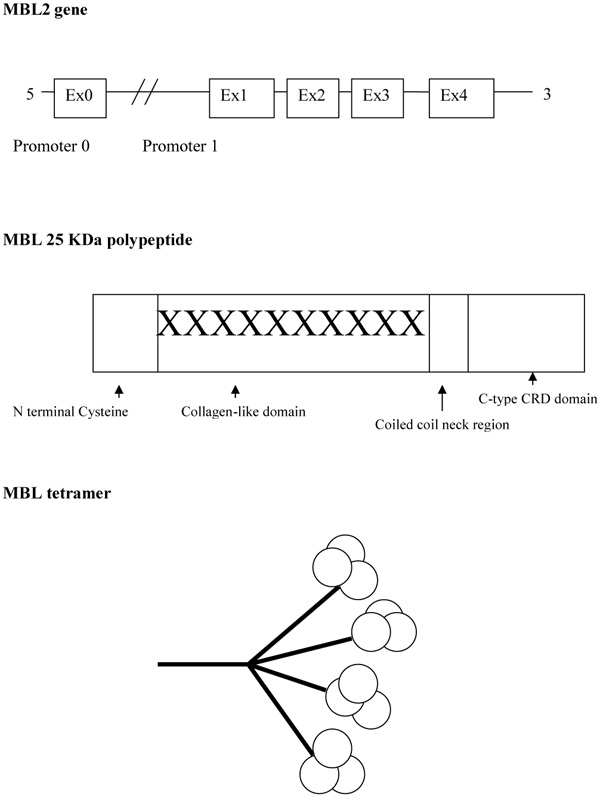
Mannose-Binding Lectin (MBL), also known as mannose-binding protein, was first discovered by Kawasaki in 1978 when a protein was extracted from rabbit liver using mannan particles from Saccharomyces cerevisiae as a probe [3]. Five years later MBL was isolated from the human liver [4]. MBL is mainly produced by the liver but low amounts have been found in the small intestine and testis tissue [5]. Lectins that are dependent upon the presence of calcium ions are named C-type lectins. MBL belongs to the class of collectins in the C-type lectin superfamily. It circulates throughout the body and is able to recognise a wide variety of common pathogens by repeating carbohydrate sequences present on microbial surfaces. MBL has an oligomeric structure (400-700 kDa), built of subunits that contain three identical polypeptide chains of 25 kDa each [6, 7]. Each polypeptide consists of a cysteine-rich N terminal region, a collagen-like region, a hydrophobic neck region, and a carbohydrate recognition domain (Fig. 2). The collagenous regions of three such chains interact to give a classical triple helix. MBL in serum primarily consists of trimers and tetramers of 9 and 12 polypeptides respectively, but the oligomers can range from dimers to hexamers [6]. These higher-order oligomers are essential for the function of MBL [8].
MBL binds carbohydrates through the c-terminal Carbohydrate-Recognition Domain (CRD) [9]. CRD is able to form bonds with hydroxyl groups on specific ligands, including mannan, N-acetylglucosamine and glucose [10]. These carbohydrates are found on pathologic microorganisms, such as bacteria, fungi, parasitic protozoans and viruses. The CRD also recognises molecular structures of dying host cells, including nucleic acids and the metalloproteases [6]. Thus MBL acts as a recognition molecule for Pathogen-Associated Molecular Patterns (PAMPS) which play a role in the initiation and regulation of the immune response [11]. On the other hand, carbohydrates that are found on mammalian glycoproteins, such as D-galactose and sialic acid, have no affinity for MBL protein. MBL protein is, therefore, able to distinguish self from non-self in initiating the innate immune pathway.
2. MBL2 GENE
There are two human MBL genes. MBL-1 is a pseudogene and only MBL-2, which comprises four exons, encodes the protein product MBL [12]. The relative efficiency of MBL function for an individual is largely determined by polymorphisms within this MBL2 gene [13]. There are three point mutations or Single Nucleotide Polymorphisms (SNPs) within the first exon of MBL2 leading to MBL variant proteins [14]. They are designated ‘B’, ‘C’ and ‘D’, in contrast to the normal or wild-type, which is designated ‘A’. Allele B represents a point mutation at codon 54 causing substitution of a glycine with an aspartic acid (GGC to GAC) [14]. Allele C represents a point mutation in codon 57 causing a glycine to be substituted with a glutamic acid (GGA to GAA) [15]. Allele D represents a point mutation in codon 52 causing an arginine to be substituted with a cysteine (CGT to TGT) [16]. Allele ‘B’, ‘C’ and ‘D’ are collectively referred to as ‘O’. Generally, wild-type A/A is associated with the highest MBL levels, A/O with intermediate and O/O with low or absent MBL. Within any given population there are different functional levels of circulating MBL, due to the differences in the frequency of MBL 2 genetic polymorphisms. B allele is frequent among Caucasians, South Americans and Asians; C allele is only frequent among Africans but is rare elsewhere, and D allele has low frequency among all populations [17]. Among normal British Caucasians, the prevalence of A/A was 60%, A/B was 21%, A/C was 5%, A/D was 10% and O/O was 4% [17].
The MBL variant proteins are unstable and probably have a shorter half-life in the circulation [18]. They have a lower molecular weight and do not effectively bind mannan or activate complement. They interfere with the formation of higher order oligomers [19]. This genetically determined MBL deficiency status influences the susceptibility, and the course of the disease, of numerous types of infectious, autoimmune, neoplastic, metabolic and cardiovascular diseases, although the conclusion is still a subject under discussion [6].
3. MBL AND ITS ROLE IN LIVER DISEASE
In a large normal population of 566 blood donors, the median serum MBL level was 1.3µg/ml (range 0-8.4µg/ml) with 10% having a very low MBL level (<0.1 µg/ml) [20]. Studies have shown that liver cirrhosis does not seem to have any impact on the serum MBL level as 10.7% cirrhotic patients were also found to have very low levels of MBL (<0.1 µg/ml). This was confirmed in a larger Hungarian study [21], which found that among patients with liver cirrhosis of various aetiologies, the median MBL level was 1.118 µg/ml, and was not statistically different compared to the healthy control (1.027 µg/ml). Analysing subsets of liver disease did not reveal any particular liver disorder with lower MBL level. However, in cirrhotic patients, the risk for infection was significantly higher among MBL deficient patients and there is a shorter time to develop infectious complications when compared to non-deficient MBL cirrhotics.
In patients with Acute Liver Failure (ALF) low levels and poor functionality of the complement system has been known for some time [22] leading to poor outcomes as patients are more prone to infection and systemic sepsis. Recently it has been shown that there are significant changes in the lectin levels of patients with ALF and furthermore high MBL levels were associated with survival [23].
In contrast, activation of the MBL/MASP activates the complement system leading to a pro-inflammatory state resulting in deposition of excess fibrogenic extra-cellular proteins resulting in fibrosis. Data in linking MBL and liver fibrosis is limited. Nevertheless, it has been shown that among HCV patients, those with severe liver fibrosis had significant higher mean serum MBL level compared to those with mild fibrosis [24], which is thought to be due to the increased complement activation as well as the increased activity of the pro-inflammatory MBL/MASP1 complexes Recombinant MASP-1 has been shown to stimulate quiescent human stellate cells to differentiate into their active state. The cytokines produced subsequently increase extracellular matrix that is important in the pathogenesis of liver fibrosis [25, 26]. Similarly, higher serum levels of MBL have been found in patients with advanced periportal fibrosis due to schistosomiasis [27].
4. MBL AND HEPATITIS B VIRUS (HBV)
Chronic Hepatitis B Virus (HBV) infection affects an estimated 240 million people worldwide and leads to a broad spectrum of liver diseases, ranging from asymptomatic carriers to self-limiting acute hepatitis, chronic infection, fulminant hepatic failure, liver cirrhosis, and hepatocellular carcinoma [28]. In a study of Chinese patients with compensated cirrhosis compared to normal controls, codon 54 mutation of MBL was associated with progression of disease in chronic hepatitis B infection and the development of spontaneous bacterial peritonitis [29]. Other studies have shown that in HBV infection, patients with MBL polymorphism resulting in low levels of circulating MBL are significantly more prone to viral persistence, the occurrence of cirrhosis and hepatocellular cancers, HBV acquisition, and a higher mortality in HBV associated ALF [30-34]. These findings, therefore, may indicate that MBL has a protective role against HBV infection. However, there are some studies which contest these findings [35, 36].
With a mother to child transmission rate of HBV in the region of 5 -10%, intra-uterine transmission of HBV may be related to a failure of HBV infected mothers to stimulate an increase in foetal MBL levels to combat the virus. It has been shown that non-infected neonates of HBV mothers have an induced increase in MBL levels conferring protection whilst neonates born with HBV have a significantly lower level of MBL [37].
5. MBL AND HEPATITIS C VIRUS (HCV)
Chronic Hepatitis C infection (HCV) is a worldwide public health problem. Approximately 75% of infected patients fail to clear the virus leading to chronic liver disease [38]. Patients with HCV infection and either heterozygote or homozygote codon 54 mutation have a significantly increased rate of progression to chronic hepatitis or cirrhosis suggesting that MBL polymorphisms may influence the course of HCV infection [39-41]. It has also been suggested that MBL-2 gene polymorphism is associated with the risk of developing pre-transplant HCV-induced hepatocellular cancer and could be utilised in predicting hepatocellular cancer risk [42]. However, some studies show conflicting results and do not support the MBL deficiency progression theory [20, 43, 44].
Response to interferon-based therapies may be determined by MBL levels as it has been shown that patients with higher levels of MBL are better responders to treatment and viral clearance [39, 44].
6. MBL POLYMORPHISMS AND INFECTION POST LIVER TRANSPLANTATION
Following liver transplantation, the donor’s liver influences the MBL status of the recipient, as the liver is the primary source of MBL production. Therefore, if the recipient receives a liver with an exon 1 MBL2 variant genotype with decreased MBL level production and its suboptimal function, it is highly plausible that the recipient is at increased risk of infection post-liver transplantation. Several studies have suggested this to be the case, finding that the presence of MBL variant alleles in the MBL gene of the donor's liver, but not in the recipient, was associated with a strongly increased incidence of clinically significant infections after transplantation [45-47].
Studies have shown that recipients of MBL-deficient livers had almost a 3-fold greater likelihood of developing a clinically significant infection, particularly an increased risk of cytomegalovirus infection [46, 47]. A study has suggested that MBL-2 polymorphisms is involved in the development of acute cellular rejection post liver transplant in HCV positive recipients, but no other studies have been performed to observe if MBL polymorphism plays a role in non HCV cellular rejection [48].
However, not all studies showed the same association. A large single-centre study involving 290 donor livers failed to find any association between the donor MBL genotypes and the risk of bacterial infection after liver transplantation [49]. Cevera et al. concurred with this finding but concluded that although liver transplantation from an exon 1 MBL 2 variant genotype was not associated with a higher incidence of infection, the reduced levels of MBL contributed to poorer outcome as infectious events were of higher severity. They proposed that MBL2 genotyping be performed as deficiency could represent a major risk factor for infection-related mortality [50].
7. POTENTIAL REASONS TO ACCOUNT FOR CONFLICTING MBL2 ASSOCIATION STUDIES
The role of MBL in the susceptibility to various diseases, including liver diseases, is sometimes controversial. For the same disease, association, or the lack of it, have been reported.
Potential reasons to explain the conflicting results in MBL2 association studies are the techniques used for MBL2 genotyping, the size of the samples studied, and the choice of controls [51, 52]. With regards to MBL2 genotyping techniques, there are currently several genotyping methods, such as multiplex PCR [53], real-time PCR with fluorogenic probes [54], and melting temperature assay [55], which are not entirely similar. Therefore there is a suggestion that a blind double check on randomly chosen samples with direct sequencing should always be considered in this kind of study [51], to ensure reliability and reproducibility of the results. With regards to the choice of control, the selection of the control population as well as the possibility of replicating the results in other ethnic groups, is a crucial point in MBL2 association studies, since the frequencies of MBL2 polymorphisms show some variability within different populations [17].
8. THE ROLE OF MBL REPLACEMENT THERAPY IN LIVER DISEASES
While there are some phase 1 [56-58] and phase 2 studies [59, 60] to demonstrate that MBL replacement therapy is safe and well tolerated, its clinical role has not yet been defined and it remains a research tool to date. Additionally, the lack of a well-established association between MBL deficiency and liver diseases does not suggest that there is a potential role for such replacement therapy.
CONCLUSION
Although the data is conflicting and no clear-cut association can be concluded between MBL polymorphisms and numerous liver diseases including liver fibrosis, viral hepatitis B, C and infection post-liver transplantation, the balance of opinion would be that MBL levels do contribute to disease progression and outcomes. Although plausible, MBL replacement therapy, which is still in its infancy stage, is unlikely to play any significant role in liver diseases. Further larger studies are required before MBL polymorphisms can be accepted, or refuted, as having an association with liver diseases.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

