All published articles of this journal are available on ScienceDirect.

Early-onset of Frontotemporal Dementia and Amyotrophic Lateral Sclerosis in an Albanian Patient with a c.1319C>T Variant in the UBQLN2 Gene
Authors Info & Affiliations
Abstract
Background:
Frontotemporal Dementia (FTD) is the second most common cause of dementia under 65 years of age; it has a prevalence of 4-15 per 100,000 persons. The overt disease usually manifests in the sixth decade, and it is extremely rare to find affected patients in their twenties.
Objective:
Here, we present the clinical and molecular genetic findings of an Albanian family with a patient with early-onset FTD and Amyotrophic Lateral Sclerosis (ALS).
Methods:
Given the great variability of clinical presentation of FTD and the number of genes involved, targeted Next Generation Sequencing (NGS) was used to screen the DNA of the 27-year-old male patient. Segregation analysis was performed in available family members.
Results and Discussion:
A variant, consisting of a proline-leucine amino acid substitution in position 440, was identified in the UBQLN2 gene on the X-chromosome. This variant was previously reported as a variant of unknown significance in a 30-year-old female patient with amyotrophic lateral sclerosis. With the description of our case, we add evidence on its involvement, also in ALS-FTD. The variant is in a functional domain important for interaction with HSP70 and this, in turn, may impair the shuttling of proteins to the proteasome leading to an accumulation of protein aggregates. The variant was inherited from the unaffected mother, in line with the fact that incomplete penetrance has been widely described for this gene.
Conclusion:
The present report adds information regarding one of 34 variants in the UBQLN2 gene reported so far in association with neurodegeneration and proposes a molecular pathogenesis of ALS-FTD in this patient.
1. INTRODUCTION
Frontotemporal Dementia (FTD) is a heterogeneous group of neurodegenerative dementias characterized by degenerative atrophic alterations of the frontal and temporal lobes of the brain [1]. It is the second most common cause of early-onset dementia and has a prevalence of 4-15 per 100,000 persons under 65 years of age, similar to that of Alzheimer’s disease [2]. The overt disease usually manifests in the sixth decade, although the age of onset can vary from the third to the ninth decade [3].
The disorder has a substantial genetic component with predominantly autosomal dominant inheritance (10-30% of monogenic FTD), and is sporadic in other cases [4]. A family history of dementia or related neurological and/or psychiatric illness is reported in 40-50% of patients with FTD.
About 50 genes have been associated with FTD and FTD-spectrum disorders [5]. The three genes most commonly implicated are microtubule-associated protein tau (MAPT) and progranulin (GRN) in less than 10% of cases each, and chromosome 9 open-reading frame 72 (C9orf72) in more than 10% of cases [6]. The fourth most common gene is TBK1, found in 1-2% of all cases [7, 8]. Variants are more rarely seen in other genes.
Although FTD is considered an adult-onset neurodegenerative disease distinct from amyotrophic lateral sclerosis (ALS), it has a spectrum, often with overlapping clinical, genetic and pathology findings. Indeed, previous molecular genetic studies in neuropathology have discovered many biological foundations for clinical coexistence of FTD and ALS. This implies variants in other genes, such as TARDBP, UBQLN2, FUS and VCP [9]. Variants considered to be risk factors for early-onset dementia may affect APP (10-15% of cases), PSEN1 (20-70% of cases) and PSEN2 (rare, approximately 5% of cases) [10].
Here, we report the case of a 27-year-old male Albanian patient with early-onset frontotemporal dementia and amyotrophic lateral sclerosis who underwent genetic testing of DNA extracted from blood. We used next generation sequencing (NGS) to formulate a molecular diagnosis. Segregation of the variant in available family members was performed to interpret the variant and predict the outcome in family members.
2. MATERIALS AND METHODS
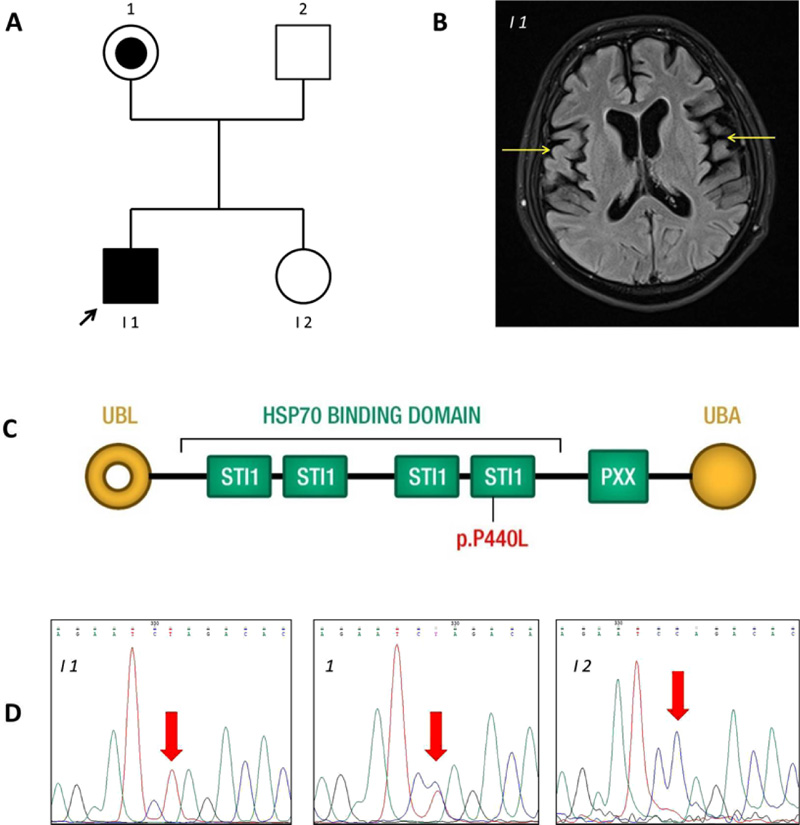
All studies on human subjects were performed according to the Declaration of Helsinki criteria and with approval from the Ethical Committee of Azienda Sanitaria dell’Alto Adige. The patient (I-1 in Fig. 1A) of the Caucasian race, with clinical signs of frontotemporal lobe dementia, was examined in the MAGI Balkans laboratory. The patient led a normal life until age 22. He was enrolled at University and practised sports. On turning 22, the first symptoms of dysphagia and difficulty walking and speaking occurred. He started to show a lack of attention and motivation. The diagnosis of ALS-FTD was made at age 24. Indeed, magnetic resonance imaging showed evident atrophy of the right and left temporal lobes (Fig. 1B) and C5-C6 cervical disc herniation (data not shown). No obvious bone lesions were detected. There are no similar cases among relatives. The patient was hospitalized for respiratory problems for seven months, where he died at age 29. Since the early onset suggested a genetic cause, a blood sample was obtained for DNA sequencing.
Genetic analysis was performed with informed consent of the patient on DNA extracted from whole blood. Targeted sequencing was performed using the Illumina TruSight One Sequencing Panel on an Illumina MiSeq platform. This kit provides comprehensive coverage of the coding regions of 4813 disease-associated genes and the respective clinical phenotypes (http://www.illumina.com/products/TruSight -one-sequencing-panel.ilmn). In-solution target enrichment was performed according to the manufacturer’s protocol. Briefly, 50 ng genomic DNA was simultaneously fragmented and tagged by Nextera transposon-based shearing technology. A 3-plex sample library pool was sequenced using a 150 bp paired-end reads protocol on a MiSeq sequencer (Illumina, San Diego, CA). We focused on 22 genes described as associated with frontotemporal dementia, according to Orphanet (http://www.orpha.net/).
Interpretation of sequence variants and reporting of results were generally implemented according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology [11, 12]. The genes analyzed are indicated in Table S1.
Bioinformatic analysis of the data was performed using an in-house pipeline to align the sequence reads with a reference genome. Variant calling, annotation and variant filtering were applied to remove benign single nucleotide polymorphisms (variants with allele frequency > 0.03 were excluded from analysis). Public databases such as those of the 1000 Genomes Project (http://www.internationalgenome.org/), Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC) (exac.broadinstitute.org/) and Single Nucleotide Polymorphism (dbSNP, www.ncbi. nlm.nih.gov/SNP/) were used to filter and prioritize the variants and to check for allele frequencies, while the Human Gene Mutation Professional Database (HGMD) (http://www.biobase-international.com/product/hgmd) was used to check for previous reports of the genetic variations. In silico prediction software such as SIFT (Sorting Intolerant from Tolerant), PolyPhen-2 (Polymorphism Phenotyping v2), Mutation Taster, Mutation Assessor and LRT (Likelihood Ratio Test) were used to assess potential deleterious effects of missense variants.
The variant detected was confirmed by PCR amplification and bidirectional Sanger sequencing of the target regions on a CEQ8800 Sequencer (Beckman Coulter) according to the manufacturer’s instructions, using the UBQLN2 gene primer sequences GTAAAACGACGGCCAGTAGCACTGGTAG TG GGTCTGG and CAGGAAACAGCTATGACCAG GGCC AGTGGGTCCTATG. Sanger sequencing was also scheduled for other family members (Fig. 1A).
3. RESULTS
Mean NGS coverage was 80.9X with 95.0% of regions having a coverage of at least 25X (coverage greater than 10X = 98.2%). NGS analysis detected the variant c.1319C>T or p.P440L in the UBQLN2 gene on the X-chromosome (rs763131369). The amino acid substitution P440L affects a highly conserved proline (P) residue in the fourth heat shock chaperonin-binding motif of UBQLN2 protein before the PXX binding domain (Fig. 1C) [13]. Sanger sequencing confirmed that the variant was inherited from the patient’s unaffected mother (Fig. 1D).
4. DISCUSSION
Mutations in 50 genes have been associated with frontotemporal dementia, a disease that may occur in isolated form or as part of a syndrome. Given the great variability of clinical presentation of ALS, FTD or ALS-FTD and the number of genes involved, an NGS approach that simultaneously tests for genes associated with the disease spectrum is suggested to increase the success rate of genetic diagnosis compared to other more time-consuming and costly approaches. The timely identification of the specific genetic profile of a patient can help understand the disease and be useful for the clinical management of patients.
The proband carries a c.1319C>T nucleotide change in the UBQLN2 gene that translates into a residue substitution of proline 440 with leucine in a heat shock chaperonin-binding motif. Ubiquilin2 is a member of the ubiquilin family and controls the degradation of ubiquitinated proteins. Variation in this protein sequence may lead to defects in the protein degradation pathway, with abnormal protein aggregation and neurodegeneration [14-16]. Ubiquilin contains a ubiquitin-like domain at the N-terminus and a ubiquitin-associated domain at the C-terminus, separated by a central region containing Sti1-like repeats. The variant p.P440L is in the fourth heat shock chaperonin-binding motif, known as the STI1 domain (Fig. 1D). Part of the central region of UBQLN2 contains domains with homology to a heat shock binding protein called STI1, which binds Stch, a protein similar to Heat Shock Protein 70 (HSP70) [17]. Under non-stress conditions, UBQLN2 associates with other UBQLNs (UBQLN1, UBQLN2 and UBQLN4), and is kept inactive. On proteotoxic stress, UBQLN2 is activated and binds the HSP70-substrate complex. Ubiquitination is not required for UBQLN2 binding. UBQLN2 then shuttles this complex to the proteasome for degradation [18], as shown on the left side of Fig. (2). The functional study performed by Kaye et al. showed that the fourth STI1 repeat sequence completely loses HSP70 binding if its structure is altered, suggesting that this region is required for binding to HSP70 [17]. The interaction between UBQLN2 and HSP70 could, therefore, be important for disaggregation activity under proteotoxic conditions. Mutant UBQLN2 is defective in the clearance of aggregates [19], and this activity could also be altered in our patient in whom the variant p.P440L affects the fourth STI1 site which is involved in HSP70 binding (Fig. 2).

The variant c.1319C>T was reported by Dillen et al. in a female patient of Bulgarian gypsy origin with the onset of ALS at age 30 years, who inherited the variant from her unaffected mother (age 67) [13]. Interpretation of the pathogenicity of this variant remains ambiguous, although its identification in another family shifts the needle of the scale to a pathogenic role. Arguments in favor of its pathogenicity are: i) two different early-onset probands have been reported with the same variant; ii) the variant is located in a functional domain important for HSP70 interaction; iii) the amino acid is highly conserved; iv) the variant has a low frequency (5 mutated alleles in a total of 183347 alleles). Arguments against the pathogenic effect are: i) the variant was transmitted to both patients (now 67 and 56 years old) by an unaffected mother. Other authors have also reported large pedigrees with healthy female carriers [14]. This variable expression of disease may be explained by an X-linked dominant inheritance model, in which males are more affected than females, being hemizygotes and therefore expressing only the mutant protein. Women are more likely to be mildly affected and can have variable disease expression due to X-skewing. X-chromosome silencing or Lyonisation in females normally occurs at random, with cells expressing the mutant X ranging from 30-70% in the case of a variant. This may explain why some females with a high percentage of mutant X express disease, while others are healthy.
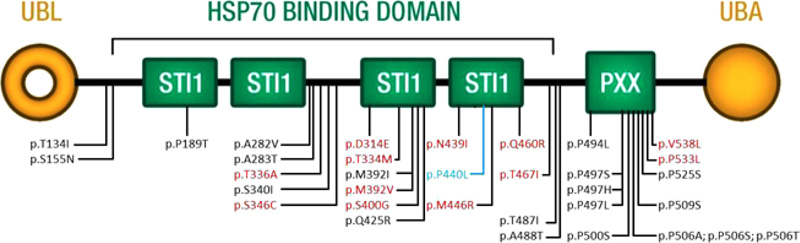
Twenty variants in UBQLN2 have been reported as disease-causing, and 14 are possibly disease-causing [14, 15, 28-37, [20-27]. Their distribution is not limited to the functional domain, as initially found by Deng and coworkers. Our variant is in the fourth domain STI1 and is the first to be identified in a male patient with such early onset (Fig. 3 and Table S2). Early onset is mainly associated with ALS, whereas in our case, it is also associated with clinical signs of FTD.
CONCLUSION
The X-linked gene UBQLN2 is involved in the pathogenesis of progressive neurodegenerative diseases, such as FTD and ALS. The present report provides more evidence regarding one of 34 variants in the UBQLN2 gene reported so far in association with neurodegeneration and proposes molecular pathogenesis of FTD in this early-onset patient. The p.P440L variant is in a functional domain important for interaction with HSP70 and this, in turn, may impair the shuttling of proteins to the proteasome, leading to the accumulation of protein aggregates, typical of these clinically heterogeneous diseases.
The data has not been published or submitted for publication elsewhere. All the authors approved the submission of the manuscript and have no conflict of interest to disclose. Informed consent was obtained from all subjects.
AUTHORS' CONTRIBUTIONS
EM, NC and MBe conceived the study; MB and EM conducted the experiments; MB, EM, PEM analyzed the data; MB and EM wrote the manuscript; NC, LB, SM, RC, BA, MBe revised the data; MBe obtained funding; all the authors read and approved the final version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval and clearance were received from the Ethical Committee of Azienda Sanitaria dell’Alto Adige, Italy (Approval No. 94-2016).
HUMAN AND ANIMAL RIGHTS
No animals were used in this research. All human research procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation (institutional and national), and with the Helsinki Declaration of 1975, as revised in 2013.
STANDARDS FOR REPORTING
CARE guidelines and methodology were followed.
CONSENT FOR PUBLICATION
Informed consent was obtained from every participant prior to the study.
FUNDING
The work was supported by funding from the Provincia Autonoma di Trento under project LP6/99 (dpg 1045/2017).
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicial to the impartiality of the reported research.
ACKNOWLEDGEMENTS
We thank the patient and his family for their collaboration. We thank all the laboratory staff of MAGI for discussion and Helen Ampt for revising the English.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.
Table S1: List of genes tested by NGS. Abbreviations: AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; PD, Parkinson’s disease; EOD, early onset dementia.
Table S2: List of 34 UBQLN2 variants identified and reported in the literature and in HGMD (www.hgmd.cf.ac.uk).

