All published articles of this journal are available on ScienceDirect.

A BRCA1 Frame Shift Mutation in Women of Kurdish Jewish Descent
Authors Info & Affiliations
Abstract
Hereditary cancer comprises more than 10% of all breast cancer cases. In patients with a family history suggestive of a hereditary component, a mutation is often identified in the high penetrant genes BRCA1 and BRCA2. Several founder mutations have been detected in some Jewish communities, yet no BRCA1/2 founder mutation had been known in Kurdish Jews. Here, we describe the validation of a 22 hereditary cancer gene panel and a BRCA1 mutation found in 4 women from 2 unrelated Kurdish Jewish families utilizing this gene panel.
A panel spanning the coding sequences of 22 familial cancer-related genes was planned. Genomic DNA was taken to create libraries using this panel, which were then sequenced using the Ion Torrent PGM. The panel's validity in detecting mutations was tested on 25 samples with previously identified point mutations in the BRCA1, BRCA2, MLH1 and PMS2 genes; the panel did not test for large deletions or insertions. All previously identified mutations were detected. Next, a different set of 40 cancer patients of Kurdish Jewish descent diagnosed with cancer before the age of 50 years was tested. We identified the BRCA1 mutation, c.224_227delAAAG (dbSNP ID rs80357697), in 4 women from 2 unrelated Jewish Kurdish families. The probands were diagnosed with cancer at a young age and had significant family history, suggesting a founder mutation in this population. We suggest testing Kurdish Jewish women with a personal or family history of breast and/ or ovarian cancer for this mutation.
INTRODUCTION
Hereditary cancer comprises more than 10% of all breast cancer (BC) cases and 20% of ovarian cancer (OC) cases [1]. In patients with a family history suggestive of a hereditary component, a mutation is often identified in the high penetrance genes BRCA1 and BRCA2 [2]. Several founder mutations have been detected in various Jewish communities in Israel [3-5]. Yet, most patients from diverse ethnic backgrounds, even Ashkenazi Jews, that seem to have a hereditary predisposition for BC/OC, appear to be non-carriers when screened just for the common founder BRCA1/2 mutations. In some of these patients, novel mutations were detected by full sequencing of the BRCA1/2 genes that later proved to be founder mutations in distinct populations, such as Sephardi Jews [4] or in Jews from Iraq, Iran and Afghanistan [6].
Up until now, no BRCA1/2 founder mutation has been known in the Jews originating from Kurdistan. The Jews of Kurdistan lived—until their immigration to Israel in the early 1950s—as a closed ethnic isolate, mostly in northern Iraq and Iran and in eastern Turkey. Genetically, Kurdish Jews are not closer to Muslim Kurds than are Sephardi or Ashkenazi Jews, suggesting that reciprocal male gene flowbetween Jews in Kurdistan and their Muslim host population was below the detectable level [7].
To identify diverse mutations in BC/OC patients, a 21-gene panel named BROCA has been utilized by other groups. In addition to sequencing the exons of BRCA1 and BRCA2, this panel also includes other high penetrance genes such as TP53, intermediate penetrance genes related to hereditary BC/OC such as PALB2, and genes involved in Lynch syndrome, as mutations in these genes increase susceptibility to OC [1].
Here, we describe the validation of a 22 hereditary cancer gene panel based on BROCA [1] with the addition of the CDKN2A [8]. Then, we describe a BRCA1 mutation (dbSNP ID rs80357697) found in 2 unrelated Kurdish Jewish families utilizing the 22-gene panel.
METHODS
Population
The panel validation included 25 patients from various origins with previously identified mutations from the cancer-genetic clinic in Hadassah Medical Center. Following the validation of the panel, 38 female and 2 male cancer patients diagnosed with cancer before the age of 50 years (30 with BC and 10 with other tumors), with either one or both parents of Kurdish Jewish descent, were also tested using the 22-gene panel. After the BRCA1 mutation had been identified, 53 additional Kurdish Jewish cancer patients and 322 cancer patients from other ethnic backgrounds, diagnosed at various ages were tested for this mutation by high resolution melting (HRM). All patients included in this study had previously signed an informed consent.
Identification of Mutations in 22 Hereditary Cancer Genes
Using the custom Ion Ampliseq panel service, a panel spanning the coding sequences of the genes ATM,BARD1,BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, CDKN2A, PALB2, PMS1, PMS2, PTEN, RAD50, RAD51C, STK11 and TP53 was planned. The panel spans between 80%-100% of the exons per gene with 96% of the bases sequenced (Supplementary Table 1). DNA extracted from the blood of consenting patients was acquired and the DNA concentration was measured using the Qubit Fluorometer. 10 ng of DNA was used in a PCR reaction which amplified exons of the 22 genes listed above using a custom primer mix. Primers were then digested, barcodes and adaptors were ligated onto the amplicons, and finally, the libraries were amplified. The libraries were then sent for mass parallel sequencing on the Ion Torrent PGM machine, aligned against the human genome hg19, the results analyzed using the Ion PGM variantCaller v4 plugin and visualized with the Integrative Genomics Viewer (IGV) software.
Sanger Sequencing
PCR primers were planned using the PRIMER3 program; they are 5'-GGTTTTCTACTGTTGCTGCATCT-3'; and 5'-TGGACAGCACTTGAGTGTCA-3'. Amplification conditions were 94° for 2 min; and 30 cycles of 94° for 30 sec, 58° for 15 sec, and 72° for 15 sec; The purified PCR fragments were sequenced by the dideoxy terminator cycle–sequencing method with AmpliTaq DNA polymerase, FS (ABI Prism Ready Reaction Kit), and then were analyzed by use of an automatic DNA sequencer (ABI).
High Resolution Melting (HRM)
Genomic DNA was analyzed using a modified HRM protocol [9]. A PCR reaction was carried out in a total of 10 µl volume using 2 µl of genomic DNA (20 ng total), 1 µl of primer mix (containing 1400 nM of both forward and reverse primers flanking the mutation site), 5 µl of Biorad SsoFast™EvaGreen®Supermix and 2 µl of water. A Rotor Gene 6000 Real-Time PCR Machine was used with the following protocol: activation step at 98°C for 3 minutes followed by 45 cycles of denaturation at 98°C for 20 seconds, and annealing at 58°C, for 20 seconds. The PCR amplicons were then heated to 98°C for 1 minute and then cooled to 50°C for 1 minute. Using the SYBR Green I channel, melt curve data was then collected from 73°C-83°C at a ramping rate of 0.5°C per second. Wild type samples displayed a single peak while the mutated samples displayed a bimodal melting curve. When repeated 26 times the mutated samples peak is at an average of 78.2° (confidence interval 77.1-78.5) and wildtype at an average of 79° (confidence interval 78.8-79.2) t-test p<0.001 (Fig. 1).
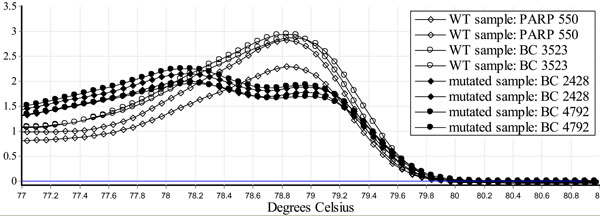
Identification of a BRCA1 c.224_227delAAAG using HRM. Genomic DNA was purified from blood of two normal controls (PARP 550 and BC 3523) and two carriers of BRCA1 c.224_227delAAAG (BC 2428 and BC 4792), using the Qiagen Flexigene DNA kit and analyzed using a modified HRM protocol [9]. When repeated 26 times, the mutated samples peak is at an average of 78.2° (confidence interval 77.1-78.5) and wildtype at an average of 79° (confidence interval 78.8-79.2) t-test p<0.001. HRM identifies the BRCA1 c.224_227delAAAG mutation.
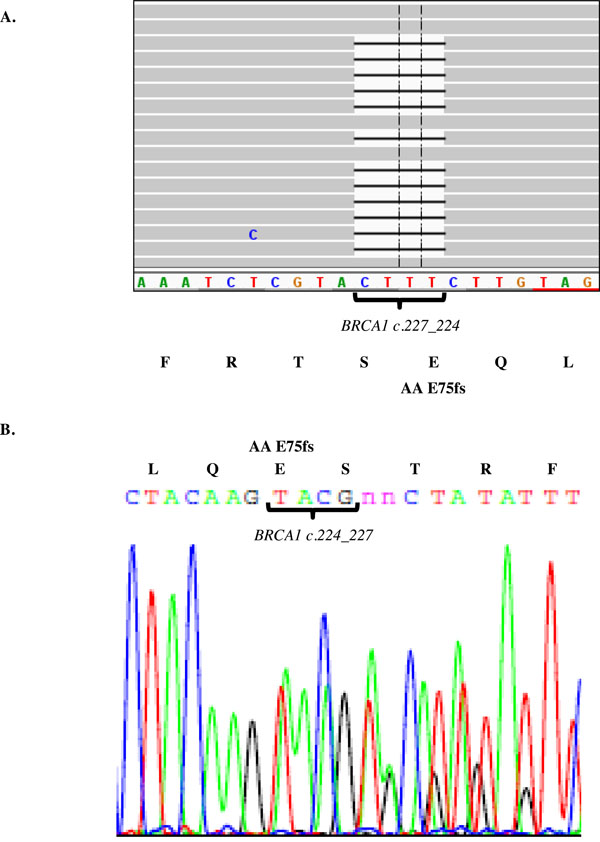
Identification of a BRCA1 frame shift mutation in Kurdish Jewish women. Genomic DNA was purified from a Kurdish Jewish women and the 22 gene panel applied. The library was massive parallel sequenced using the Ion Torrent PGM and visualized using the Integrative Genomics Viewer (IGV) software. The BRCA1 c.224_227delAAAG is identified using the panel approach (A) and verified by Sanger sequencing (B). This mutation in Kurdish Jewish women is a deletion resulting in a frame shift from amino acid 75.

Pedigree of the BRAC1 c.224_227delAAAG carriers. Four women from two unrelated Kurdish Jewish were consulted in the cancer-genetic clinic of the Hadassah medical center. The probands were diagnosed with cancer at a young age and had significant family history, suggesting a founder mutation in this population.
| BC Sample # | Gene | Position | DNA Mutation | Protein Mutation | SNP | HGMD [10] | Mutation Taster |
| 6692 | ATM | Chr11:10812355 1 | c.1810C>T | P604S | rs2227922 | DM [13] | DC1 |
| 6692 | BRCA2 | Chr13:32912750 | c.4258G>T | D1420Y | rs28897727 | DM? [14] | P2 |
| 6692 | MRE11AA | Chr11:94180441 | c.1727G>A | R576Q | rs139461096 | Not found | P |
| 4734 | ATM | Chr11:10816417 | c.4743C>G | I1581M | Not found | Not found | P |
| 388 | ATM | Chr11:10818673 | IVS41-3T>C | splice site | Not found | Not found | DC |
| 388 | CDH1 | Chr16:68855966 | c.1774G>A | A592T | rs35187787 | DM? [15] | DC |
| 388 | MSH6 | Chr2:48027683 | c.2561_2562delinsTT | K854I | Not found | Not found | DC |
| 388 | MSH6 | Chr2:48027755 | c.2785T>C | V878A | rs2020912 | DM? [16] | DC |
| 2428 | BRCA1 | Chr17:41256959 | c.224_227delAAAG | E75fs | Not found | Not found | DC |
| 2428 | PMS1 | Chr2:190742010 | c.1516C>T | R506C | rs142159998 | Not found | DC |
| 4792 | BRCA1 | Chr17:41256959 | c.224_227delAAAG | E75fs | Not found | Not found | DC |
| 4792 | CHEK2 | Chr22:29130616 | c.94T>G | S32A | Not found | Not found | P |
| 5860 | BRCA1 | Chr17:41246481 | c.1067A>G | Q356R | rs1799950 | DP [17] | P |
| 5728-1 | BRCA2 | Chr13:32911046 | c.2554A>C | N852H | Not found | Not found | P |
| 5728-2 | BRCA2 | Chr13:32911046 | c.2554A>C | N852H | Not found | Not found | P |
| 7091 | BRCA2 | Chr13:32912750 | c.4258G>T | D1420Y | rs28897727 | DM? [14] | P |
| 2362 | BRCA2 | Chr13:32914814 | c.6322C>T | R2108C | rs55794205 | DM? [18] | P |
| 7307 | BRCA2 | Chr13:32972626 | c.9976A>T | K3326* | rs11571833 | DP [19] | DC |
| 6141 | BRIP1 | Chr17:59924512 | c.577G>A | V193I | rs4988346 | Not found | P |
| 2380 | PALB2 | Chr16:23641515 | c.1960A>G | I654V | Not found | Not found | P |
| 2118 | PALB2 | Chr16:23641340 | c.2135C>T | A712V | Not found | P | |
| 6812 | RAD50 | Chr5:131977933 | c.3816T>G | D1272E | Not found | Not found | DC |
| Thyroid cancer 5144 | ATM | Chr11:108117798 | c.1009C>T | R337C | rs138398778 | DM [20] | DC |
| Rectal cancer 3912 | BRCA1 | Chr17: 41234505 | c.964C>T | P322S | Not found | Not found | P |
| 3912 | MSH6 | Chr2:48027683 | c.2561_2562delinsTT | K854I | Not found | Not found | DC |
| Uterine cancer | |||||||
| 5626 | BRIP1 | Chr17:59926582 | c.415T>G | S139A | rs202072866 | Not found | DC |
| Colon cancer 493 | BRIP1 | Chr17:59924512 | c.577G>A | V193I | rs4988346 | Not found | P |
| 493 | BRIP1 | Chr17:59820428 | c.2325T>G | N775K | rs375146450 | Not found | DC |
| 493 | CDH1 | Chr16:68855966 | c.1774G>A | A592T | rs35187787 | DM? [15] | DC |
| 493 | P53 | Chr17:7579467 | c.220G>A | A74T | Not found | Not found | P |
| Colon cancer 6532 | MSH6 | Chr2:48027755 | c.2785T>C | V878A | rs2020912 | DM? [16] | DC |
| Cervical cancer | RAD50 | Chr5: 131945093 | IVS19+5G>A | splice site | Not found | Not found | P |
Genomic DNA was purified from a Kurdish Jewish women and the 22 gene panel applied. The library was massive parallel sequenced using the Ion Torrent PGM and visualized using the Integrative Genomics Viewer (IGV) software. All mutations were validated using Sanger sequencing. Missense, nonsense, frameshift and splice site mutations that are either pathogenic, not found or found in less than 1% of the population are displayed. The prediction is based on HGMD [11] and mutation taster [12].
RESULTS
A Panel of 22 Hereditary Cancer Genes Based on the BROCA Gene Panel [1] is Valid in Detecting Point Mutations
Non-synonymous mutations and indels in coding regions were identified in 23/25 samples, including all carriers of BRCA1 185delAG (n=5); BRCA1 5382insC (n=3); BRCA1 E1373X (n=1); BRCA1 A1708E (n=1); BRCA1 3053T-G (n=1); BRCA1 3832C>T (P1238L) (n=1); BRCA2 6174delT (n=3); BRCA2 8675delAG (n=1); BRCA2 IVS2+1 G>A (n=1); BRCA2 1153insT (n=1); BRCA2 6024dupG (n=1); BRCA2 969 C>T (n=1); MLH1 655 A>G/N (n=1); PMS2 943 C>T (n=1) and PMS2 2192 T>G (n=1). The BRCA1 Ex 18-20 dup (n=1) and BRCA2 Del Ex 12-13 (n=1) mutations were not detected.
Identification of a BRCA1 Frame Shift Mutation in Kurdish Jewish Women
Using a different cohort than the one used for panel validation, 40 Jewish Kurdish patients were tested using the 22 gene cancer panel, mean read depth 228. In 2 out of the 39 unrelated Jewish Kurdish patients tested, a common mutation was identified in the BRCA1 gene. This mutation is a CTTT deletion in hg19 chr17:41256959 resulting in a frame shift, c.224_227delAAAG, from amino acid 75, and was verified using Sanger sequencing (Fig. 2). The mutation, dbSNP ID rs80357697, has been described in a two cases with no ethnic data. It causes a frame shift that is expected to disrupt part of the RING domain, two nuclear localization sequences, binding sites for RB, cMYC, RAD50 and Rad51, a serine cluster domain and a BRCT domain [10]. In one family, the proband (patient sample number 2428) suffered from triple negative breast cancer at the age of 36, and her mother suffered from fallopian tube cancer at the age of 38. Two maternal aunts suffered from ovarian cancer at the age of 45, and one maternal aunt suffered from breast cancer at the age of 60. In the second family, the proband (patient sample number 4792) suffered from ER positive breast cancer at the age of 40. Her mother (patient sample number 509) suffered from a medullary carcinoma of the breast at the age of 59 and synchronous triple negative breast cancer and lung cancer at the age of 74. Four maternal cousins suffered from cancer. Using HRM and Sanger sequencing, the mother (patient sample number 509) and a maternal cousin's daughter (patient sample number 687) were found to be a carriers of the BRCA1c.224_227delAAAG mutation, (Fig. 3). No additional carriers of the BRCA1c.224_227del AAAG mutation were identified among the 53 cancer patients of Kurdish Jewish descent or 322 cancer patients from other ethnic backgrounds who were screened using HRM.
Other mutations identified using the 22 gene panel and verified using Sanger sequencing in the 40 study patients included missense, nonsense and splice site mutations in the ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MRE11A, MSH6, P53, PALB2, PMS1 and RAD50 genes. Based on the Human Gene Mutation Database (HGMD) [11] and mutation taster [12], some of these mutations are polymorphisms while others are disease causing (Table 1).
DISCUSSION
We report the dbSNP ID rs80357697 cancer susceptibility mutation in the BRCA1 gene in 4 women from 2 families of Kurdish Jewish descent. The personal and family history of the patients is typical to BRCA1 mutation carriers, including women affected with breast cancer at a young age and women with ovarian or fallopian tube cancer. Finding this described mutation in two unrelated families from the same origin suggests that this mutation is a founder mutation in the Kurdish Jewish community. Yet, proof of this assumption would need haplotype comparison between the two families, which could not be performed because, unfortunately, relatives were not available.
The mutation was detected in two out of thirty women diagnosed with breast cancer under 50 years of age, and was not demonstrated in 53 unselected cancer patients of Kurdish Jewish origin or in 322 unselected cancer patients from other ethnic backgrounds. We plan to extend out cohort and test healthy controls to gain more information about the allele frequency of this mutation. We intend to screen for this mutation in our future patients and suggest other genetic centers in Israel to do as well. By early detection of mutations in healthy probands, preventive treatment may be offered and BC/OC morbidity minimized [2]. Identification of this mutation in ovarian cancer patients can offer novel treatment opportunities, such as treatment with PARP inhibitors [13].
To conclude, we suggest testing for the mutation BRCA1c.224_227delAAAG in Kurdish Jewish women with a family history of BC/OC. Panel screening of patients from other specific ethnic backgrounds may identify other founder mutations.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGMENTS
This research was supported by THE ISRAEL SCIENCE FOUNDATION (Grant No. 1985/13).
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.

